Mišićno koštani tumori
 |
Udruga Ljubav Na Djelu | | 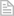 |
27.07.2024 |
Mišićno koštani tumori
Dijele se na :
Rak kostiju:
Kost ili koštano tkivo jest povezivno tkivo koje podupire tjelesnu strukturu. Sve kosti u ljudskom tijelu zajedno čine ljudski kostur te zajedno s mišićima kosti čine sustav organa za kretanje.
Kosti su međusobno povezane pokretljivim zglobovima i mišićima tako da mogu pokrenuti tijelo brzinom do 45 km/h. Kosti su čvrste i jake, šuplje unutrašnjosti, stoga nisu teške te čine samo 14% naše ukupne tjelesne mase (lakše su od mišića). Kosti nisu mrtve tvari u našem organizmu, pa se zato obnavljaju kad se prelome. Ljudski kostur ima 206 kostiju.
Građa kostiju:
- Čvrsto koštano tkivo prožeto je kanalićima oko kojih su kružno poredane koštane stanice, koje u međustanični prostor izlučuju kalcij i fosfor što kostima daje čvrstoću. Kanalićima prolaze krvne žile i živci.
- Hrskavica je glatka i čvrsta savitljiva nadopuna kostima a smanjuje trenje u zglobu.
- Spužvasto koštano tkivo jest koštano tkivo ispunjeno šupljinama i išarano malim potpornjima koji čine kosti jakima ali ne i preteškima.
- Pokosnica obavija kost i čvrsto prirasta uz nju. Građena je od posebnog vezivnog tkiva.
- Koštana srž - Cjevaste šupljine dugih kostiju i šupljine u spužvastom tkivu ispunjene su koštanom srži koja proizvodi većinu krvnih stanica.
Primarni rak kostiju je rak koji se razvija u stanicama kostiju.
Vrste primarnih raka kostiju su:
- Osteosarkom: najčešći oblik raka kostiju. Najviše zahvaća mlade između 10 i 25 godina života, u razdoblju kada kosti intenzivno rastu te pogađa više muškarce nego žene. Kod djece i adolescenata najčešće se pojavljuje pri kraju dugih kostiju ruku i nogu, posebice u području oko koljena. Simptomi, kao i mogućnosti izlječenja, kod djece i odraslih su jednaki. U trenutku dijagnosticiranja 15-20% pacijenata ima uočljive metastaze na plućima ili ostalim kostima. U SAD-u godišnje se dijagnosticira oko 400 novih slučajeva osteosarkoma.
- Ewingov sarkom: razvija se u nezrelom (primitivnom) živčanom tkivu u koštanoj srži.
- maligni fibrozni histiocitom (malignant fibrous histiocytoma), sarkom mekog tkiva, vrlo rijetko se razvija u kosti. Može se javiti kao posljedica zračenja. Primjenjuje se isti način liječenja kao kod osteosarkoma i čini se da ima sličan odgovor na terapiju.
- hondrosarkom: razvija se u hrskavici.
Vrsta raka |
Podrijetlo stanica/tkiva |
Uobičajena lokacija |
Starost |
Osteosarkom |
Osteoid |
Koljena, bedra, nadlaktice |
10-25 |
Hondrosarkom |
Hrskavica |
Zdjelica, bedra, ramena |
50-60 |
Ewingov sarkom |
Nezrelo živčano tkivo (obično u koštanoj srži) |
Zdjelica, bedra, rebra, ruke |
10-20 |
Osteosarkom i Ewingov sarkom se najčešće pojavljuju kod djece i mlađih osoba do 25 godina, a hondrosarkom se najčešće pojavljuje kod starijih osoba.
Sekundarni rak kostiju je rak koji se je proširio na kosti iz nekih drugih dijelova tijela (npr. dojke, pluća, crijeva).
Osteosarkom:
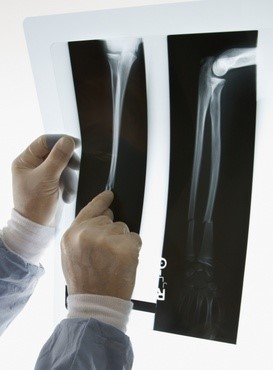
Osteosarkom je najčešći tumor kostiju u dječjoj dobi. Većinom se javlja na dugim kostima, na primjer na nadlaktici ruke i natkoljenici noge, gdje dolazi do destrukcije - razaranja površinskog sloja (periosta). Osteosarkom se javlja većinom kod mladih ljudi u dobi između 10 i 25 godine, a češći su u muških nego kod ženskih pacijenta. Pacijenti se žale na bolove, nekad otekline na oboljelim kostima koje se često greškom pripisuju nezgodama. Postavljanje dijagnoze može biti teško pošto se ova bolest može zamijeniti sa lokalnim infekcijama, posljedicama povreda, artritisom, manjkom vitamina ili sa dobroćudnim tumorom. Iako se osteosarkom može otkriti pomoću rentgenske slike, ipak se dijagnoza mora postaviti pomoću biopsije zahvaćene kosti. Pošto kod postavljanja dijagnoze bolesti mogu biti prisutne i metastaze (npr. pluća), prije početka liječenja potrebno je učiniti i pretrage pomoću CT-a.
U liječenju osteosarkoma koristi se kemoterapija, a potom operativni zahvat.
Danas se u mnogim slučajevima mogu izbjeći amputacije. U bolnici će doktori sa vama raspraviti o mogućnosti koja je za vaše dijete ispravna.
Uzroci i rizični faktori
Ne postoji jasno definiran uzrok osteosarkoma, iako postoji tendencija da se ovaj tumor javi unutar obitelji. Također ne postoje ni poznate predispozicije za razvoj osteosarkoma.
Dijagnosticiranje
Osobe kojima je kasnije dijagnosticiran osteosarkom, liječniku se javljaju sa sljedećim sipmtomima:
- bol
- osjetljivost na dodir
- natečene kosti,
- ograničeni pokreti
- iznenadan prijelom u inače zdrave osobe
Kako se dragocjeno vrijeme ne bi gubilo na pogrešne dijagnoze i načine liječenja, nužna je rendgenska snimka područja zahvaćenog osteosarkomom. Jednom kada rendgenske snimke potvrde postojanje tumora, slijede računalna tomografija (CT), skeniranje magnetskom rezonancom (MRI) ili pozitronska emisijska tomografija (PET). Također, specijalist može obaviti i biopsiju kako bi utvrdio o kakvom se tipu stanica radi. Lokacija, veličina i proširenost tumora utječu na izbor i na ishod liječenja.
Liječenje
Za liječenje osteosarkoma zaduženi su stručno kliničko osoblje, specijalisti ortopedi i onkolozi. Kemoterapija može smanjiti tumor i tretirati moguća moguća metastaziranja, primjerice na pluća. Nakon toga može uslijediti kirurška operacija ekstremiteta, odnosno zahvaćenog područja, kako bi se spriječila amputacija. Rekonstruktivna kirurgija također može biti opcija, a može uključiti niz operacija kako bi se reimplantirala tetiva ili ugradila proteza.
Skeniranje U nekim slučajevima, nakon kemoterapije nije moguća operacija. U drugima, postoje oblici operacija koji su mogući, ali oni mogu oslabiti organizam. U takvim situacijama razmatra se radijacija, posebno ako je biopsijom ustanovljeno da se radi o specifičnom tipu stanica. Treba se poprinuti i za moguće komplikacije tijekom liječenja, kao i za nuspojave poput infekciju rane, bol i mučninu.
Ishod liječenja
Ishod uvelike ovisi o tome u kojem je stadiju dijagnosticiran osteosarkom, o vještini liječnika i medicinskog osoblja, te o načinima liječenja raka. Ukoliko je bolest otkrivena relativno rano i liječena pravilno, šanse za preživljavanje prelaze 90 posto. Ako se radi o agresivnim oblicima tumora, koji su počeli metastazirati, šanse za preživljavanje iznose 50 posto. Ipak, točne prognoze se ne mogu davati jer svaki pacijent na liječenje reagira drugačije.
Ewing sarkom
Ewingov sarkom je maligni koštani tumor koji je prvi put u literaturi opisao J a m e s E w i n g 1921. godine. Ewingov sarkom se obično javlja u dječjoj ili ranoj adolescentnoj dobi, s najvećom učestalošću između 10. i 20. godine života, premda se može javiti i kod mlađe i starije životne dobi. Najučestalije se javlja u području zdjelice, natkoljenične i nadlaktične kosti te rebara. Ewingov sarkom je po učestalosti pojavljivanja drugi tumor koštanog tkiva kod djece i najmaligniji koštani tumor dječje dobi. Učestalost obolijevanja je 0.3 bolesnika na 1.000.000 djece mlađe od 3 godine i 4.6 djece na 1.000.000 mladih ljudi između 15 i 19 godina života. Prosječna učestalost obolijevanja je ispod 2 bolesnika na 1.000.000 djece na godinu. Odnos muške i ženske djece koja obolijevaju je 1.5:1. Preživljavanje kod lokaliziranog tumora je 60-70% oboljelih, 30% preživljavanje je s metastazama po plućima te preživljavanje manje od 10% je s metastazama na drugim mjestima.
James Ewing je opisao Ewingov sarkom kao tumor plosnatih i dugih kostiju osjetljiv na zračenje. Za ovaj maligni tumor s nediferenciranim malim okruglim stanicama se do 1980-ih mislilo da ima endotelijalno podrijetlo, no noviji podaci upućuju da se najvjerojatnije radi o najnezrelijoj stanici iz neuroektoderma (postganglijskih parasimpatičkih holinergičkih neurona). Mada je koštani tumor, Ewingov sarkom može izrastati iz mekih tkiva - ekstraosalni Ewingov sarkom (EES) ili se može pojaviti kao diferenciraniji oblik poznat pod imenom periferni primitivni neuroekodermalni tumor (PPNET). Zbog toga je opće prihvaćen termin "Ewing sarcoma family of tumors" (ESFT).
Tumor izaziva lokalnu bol u kostima ili otok mekih tkiva. Smješten je na dijafizama dugih kostiju (bedrene), i na plosnatim kostima trupa (kralješci, rebra, zdjelična kost). Češći je u drugoj dekadi života (75% dijagnosticira se prije 20 godine života a 90% prije 30 godine). Za razliku od osteosarkoma često postoji intermitentno povišenje tjelesne temperature, a javljaju se i drugi sistemni simptomi: gubitak na tjelesnoj težini, opća slabost i febrilitet, koji ujedno ukazuju na metastaze. Oko 25% bolesnika ima pri početku bolesti hematogene metastaze u plućima, kostima, koštanoj srži ili pleuri. Rendgenske snimke prikazuju tumor dijafiza koji se proteže do metafiza. Postoje miješani litički i slerozirajući defekti, ili lamele sa stvaranjem nove kosti. Oko ⅔ bolesnika ima i tumorsku masu koja se sastoji od mekog tkiva, a ponekad su prisutne patološke frakture.
Citogenetske i molekulske analize tumorskog tkiva pokazuju pet recipročnih translokacija od kojih je najčešća t(11;22)(q24;q12) koja rezultira fuzijom gena EWS-FLI1 - (90 do 95% tumora) a slijedi je po učestalosti t(21;22)(q22;q12) čija je posljedica fuzija gena EWS-ERG (5-10% tumora). RT-PCR-om i FISH-om moguće je otkriti specifične prijepise EWS-FLI1 i EWS-ERG koje izražava tumor. Ove će metode olakšati dijagnozu koja je do sada zbog nedostatka specifične patološke slike uglavnom temeljena prema mjestu pojave tumora i isključivanju drugih tumora malih okruglih stanica.
CT pluća i scintigrafija kosti sa tehnecijem neophodna je za traženje metastaza. Potrebna je i analiza koštane srži. Kompjuterizirana tomografija kosti na kojoj se nalazi primarni tumor daje vrijedne informacije o proširenju bolesti na meka tkiva i medularnu šupljinu, što je značajno pri planiranju operacije pri kojoj se otklanja kost uz čuvanje ekstremiteta. Naime Ewingov sarkom širi se iz kosti u kojoj nastaje preko medularnog kanala, ili probija korteks i širi se u okolno meko tkivo, infiltrira okolne mišiće, i duž fascija često prelazi i preko proksimalnog zgloba.
Preživljenje od oko 15% bilo je uz liječenje kirurškim zahvatom, a radioterapija kao izolirana metoda imala je još slabije rezultate od oko 10%. Ewingov sarkom nužno je liječiti kao sistemno oboljenje tz. kombinacijom kirurgije, zračenja i kemoterapije da se kontrolira primarni tumor, i eradiciraju udaljene metastaze. Kirurški zahvat ne smije biti suviše mutilirajući. Amputiraju se distalna fibula, tibia, talus, i kalkaneus. Tumore prstiju i malih tarzalnih i metatarzalnih kostiju liječe se hemiamputacijom stopala. Tumori rebara, klavikule i skapule se široko ekscidiraju, obično nakon preoperativne primjene citostatika. Proksimalna tibija, radijus i femur se ne amputiraju osim ako nisu jako destruirani.
Zrači se cijela dužina zahvaćene kosti sa 6000-7000 cGy no kako te doze dovode do jakih fukcionalnih oštećenja u kombinaciji s kemoterapijom mogu se smanjiti na 4000-5000 cGy bez utjecaja na konačni ishod. "VACA", "VIDE", "VAI" i slične programe citostatskog liječenja preporučuju Intergroup Ewing's Sarcoma Study (IESS) i Coperative Ewung's Sarcoma Study (CESS). EURO-E.W.I.N.G. 99. Preživljenje uz ovakvu kombiniranu terapiju kreće se od 55-65%
Rabdomiosarkom
Nastaje u ćelijama mišića. Češće se javlja kod dečaka nego kod devojčica i to najčešće u uzrastu između 2 i 6 godina.
Iako se može javiti u svakom mišićnom tkivu, većinom se javlja u predjelu glave i vrata, zdjelici i ekstremitetima. Rhabdomiokarcom veoma brzo raste i širi se.
Simptomi se srećom lakše pokazuju nego kod većine drugih vrsta tumora dečjeg uzrasta. Većinom se primeti otok ili opipljiv čvor. Ostali simptomi zavise od lokalizacije tumora. Ako raste na primjer u predjelu oka onda vid slabi. Ako je napadnuto grlo može doći do problema u gutanju.
Točnu dijagnozu osigurava biopsija tkiva napadnutog tumorom. Uz pomoć ultrazvuka, rengenskog snimka, scintigrafije, magnetne rezonance (MR), lumbalne i punkcije koštane srži, isključuju se metastaze.
U terapiji se koristi kombinacija operacije, kemoterapije i zračenja. Ako je tumor veliki ili je dao metastaze prvo se daje kemoterapija i zračenja, da se pokuša tumor smanjiti do određene veličine, da se može lagano operativno odstraniti.
Približno 60-70% djece ozdravi.






